- · 《实用肿瘤杂志》期刊栏[01/26]
- · 《实用肿瘤杂志》投稿方[01/26]
- · 实用肿瘤杂志版面费是多[01/26]
一、本刊要求作者有严谨的学风和朴实的文风,提倡互相尊重和自由讨论。凡采用他人学说,必须加注说明。 二、不要超过10000字为宜,精粹的短篇,尤为欢迎。 三、请作者将稿件(用WORD格式)发送到下面给出的征文信箱中。 四、凡来稿请作者自留底稿,恕不退稿。 五、为规范排版,请作者在上传修改稿时严格按以下要求: 1.论文要求有题名、摘要、关键词、作者姓名、作者工作单位(名称,省市邮编)等内容一份。 2.基金项目和作者简介按下列格式: 基金项目:项目名称(编号) 作者简介:姓名(出生年-),性别,民族(汉族可省略),籍贯,职称,学位,研究方向。 3.文章一般有引言部分和正文部分,正文部分用阿拉伯数字分级编号法,一般用两级。插图下方应注明图序和图名。表格应采用三线表,表格上方应注明表序和表名。 4.参考文献列出的一般应限于作者直接阅读过的、最主要的、发表在正式出版物上的文献。其他相关注释可用脚注在当页标注。参考文献的著录应执行国家标准GB7714-87的规定,采用顺序编码制。
14+NC,全代码,完整复现!实用!
作者:网站采编关键词:
摘要:用Nature Communication学画 图5(2)的画图 Nature Communication文章转载大结局 大家好,我是阿辰。在前面的讲解过程中,我们逐步演示了Celligner算法的构建,并探索和分析了校正后的数据是否
用Nature Communication学画
图5(2)的画图
Nature Communication文章转载大结局
大家好,我是阿辰。在前面的讲解过程中,我们逐步演示了Celligner算法的构建,并探索和分析了校正后的数据是否与不同的肿瘤亚型或亚型具有相同的对应关系。最后,整篇文章的再现过程进入了最终的内容,药物敏感性分析。
 接下来,我们来看看如何绘制和再现图5的剩余图片。
接下来,我们来看看如何绘制和再现图5的剩余图片。
1. R包读取
和前面的分析过程一样,我们首先通过library()函数读取对应的R包和相关的自定义函数。
二、数据准备
R包准备好后,接下来就是数据准备过程。在这里,我们首先读取在初步分析过程中准备的输入数据或输出结果。
2.1 读取临床注释数据
首先我们还是需要读取24个临床相关的注释文件Celligner_info.
结果显示:
2.2 基因注释数据
其次是基因信息文件gene_expression_of_avg&SD;从前面的分析中得到,包括基因名称及其在组织和细胞系中表达的肿瘤均值和方差值。
2.3 细胞表达数据
之后我们读取了从DepMap数据库19Q4版本下载的CCLE细胞系的表达数据和CCLE细胞系的计数数据。
结果表明:
结果表明,表达矩阵中行为细胞系的名称被列为基因名称。与有组织的表达矩阵不同,矩阵的列名包括基因符号和Entrez id。此内容需要在分析前进行简单的数据清理。
2.4 CRISPR敲除数据
另外,CRISPR敲除数据Achilles_gene_effect表示使用敲除基因后产生真正消费效应的基因效应的概率。 Achilles 数据集包含 689 个细胞系中 18,333 个基因的基因组规模 CRISPR 敲除筛选结果。
结果显示:
2.5?药敏数据
之后,我们进一步从DepMap数据库下载了药敏相关的数据内容。其中,PRISM Repurposing 版本包含使用 Broad Repurposing 库和 PRISM 多细胞系活力测定生成的小分子活力数据集,PRISM Repurposing 数据集包含针对 578 个细胞系筛选的 4,518 种化合物的化学扰动活力筛选。结果。
结果显示:
最后,这是细胞系对应的相关样本信息。如您所见,它包含 26 种不同的信息内容。
结果显示:
 三、图? 5E再现
三、图? 5E再现
接下来,我们来看看如何再现图5E。
3.1?图5E输入数据的准备
首先,定义感兴趣的集群,即第12个集群和第28个集群。其次,对单元格行计数表达式数据CCLE_count的列名进行拆分排序。
随后筛选了来自细胞系中第12和28个簇的样本id信息。结果表明,最终得到252个样品。
同时提取注释信息中细胞系对应的组织源covars和临床信息cell_line_info,根据细胞表达矩阵中的基因顺序对基因注释文件gene_info进行重新排序。
最后使用edgeR包和limma包对细胞系表达计数数据进行差异表达分析。
结果显示:
到此,图5E的输入数据就基本准备好了。
3.2?图5E的再现
接下来开始图5E的绘制过程。
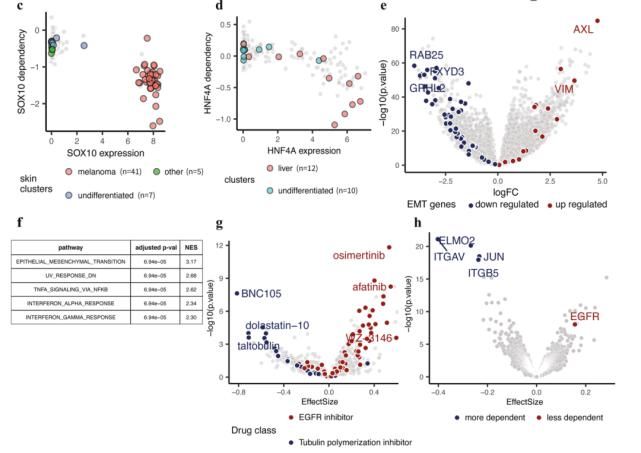
首先,由于作者在研究EMT相关通路,我们需要在差异分析结果中标注EMT通路相关基因。
使用ggplot()函数将差异分析结果可视化。可以看到,这其实是我们平时使用的火山图绘制流程的升级版。它可以标记任何感兴趣的基因,值得学习。
结果显示:
 4.图5F的再现
4.图5F的再现
接下来,这是对图5F的分析和绘制,??即GSEA分析。
首先,和常规分析一样,从差异分析结果列表中提取logFC结果,形成一个新的向量gene_stat。
其次,从msigdbr包的msigdbr()函数中提取C2基因集的相关基因信息。
然后是GSEA分析过程。使用fgsea()函数进行GSEA分析,最终得到186个通道对应的分析结果。
结果展示:
得到分析结果后,得到前5个结果,使用fgsea包的()函数绘制表格。
结果显示:
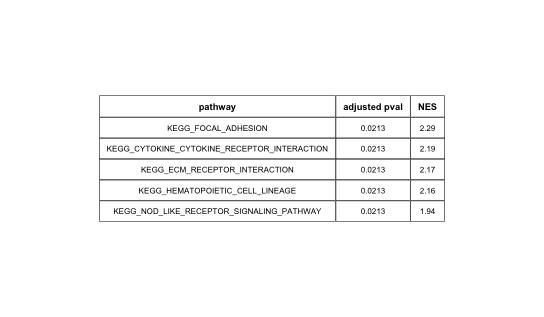 这样就完成了GSEA富集分析和表格绘制.当然,由于输入基因集的不同,最终的分析结果与原文有些不同。
这样就完成了GSEA富集分析和表格绘制.当然,由于输入基因集的不同,最终的分析结果与原文有些不同。
5.图5G再现
5.1 图5G输入数据准备
首先,根据样本信息文件中细胞系CCLE_Name的名称,对药敏行名称进行重新组织命名数据集。
然后,提取药物浓度为2.5uM的相关信息。
结果展示:
结果展示:
结合使用group_by()函数和summarise_all()函数对结果进行统计分析药品名称,同时去掉NA数据。
此外,我们还需要进一步将数据集中的缺失或NA数据内容转化为0。
另外,获取细胞系名称shared_cell_lines和复合信息cpds_to_test作为最终分析。
文章来源:《实用肿瘤杂志》 网址: http://www.syzlzz.cn/zonghexinwen/2021/0727/1215.html